Trimming Sequences
Before aligning sequencing reads, it’s important to remove adapter sequences and low-quality bases.

Read more about Illumina adapter sequences
Cutadapt
Cutadapt is a program that finds and removes adapter sequences, primers, poly-A tails and other types of unwanted sequence from your high-throughput sequencing reads.
Essentially, Cutadapt trims adapters from short read Illumina data.
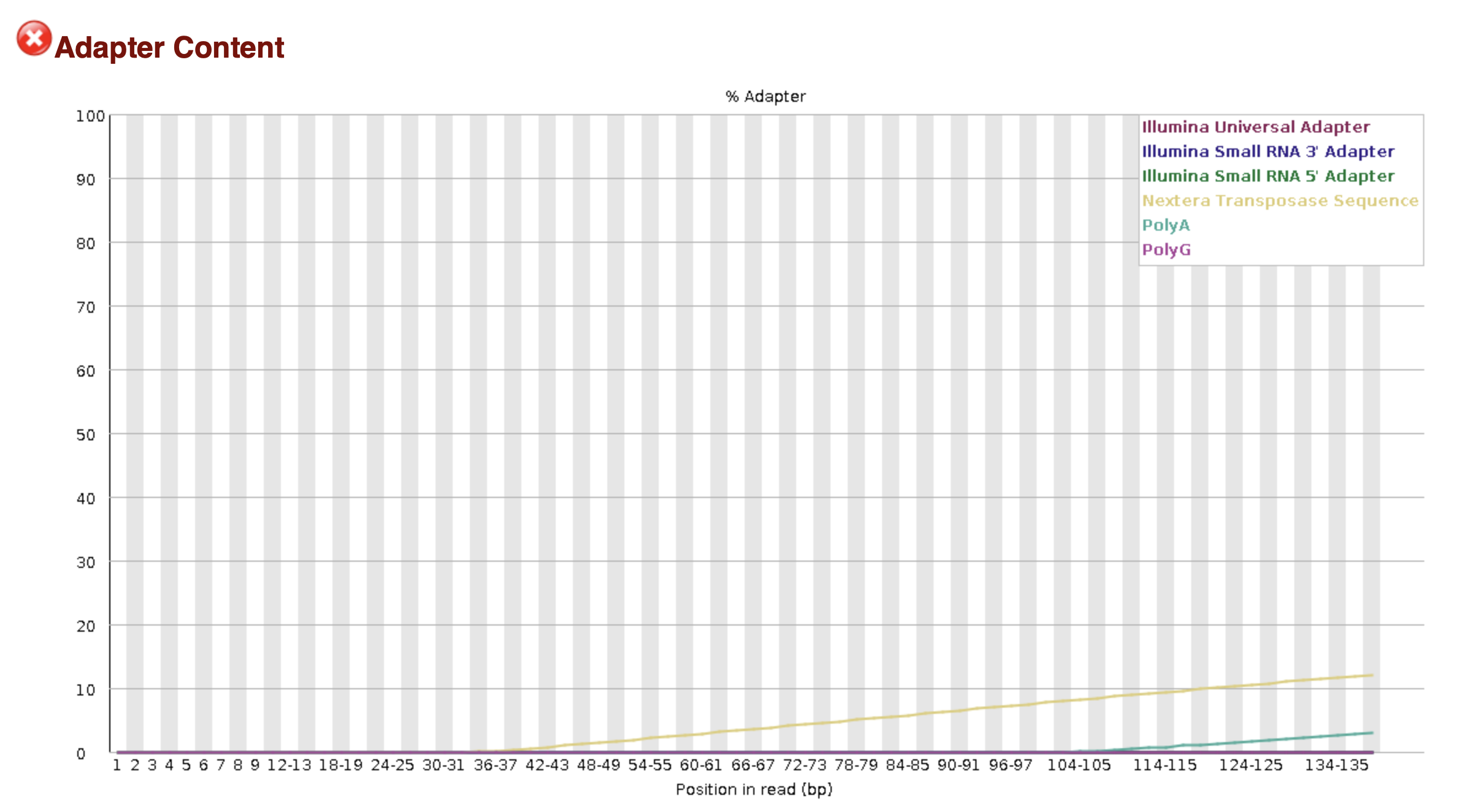
The above image shows a FASTQC .html report indicating that adapter sequences are still present in the reads. In this case, you would use Cutadapt to trim the adapters.
Using Cutadapt
Check available versions on the cluster:
$ module spider cutadapt
Output:
cutadapt: cutadapt/4.9
This module can be loaded directly:
module load cutadapt/4.9
Sample run command:
$ cutadapt -a CTGTCTCTTATACACATCT -o SRR2584866-trimmed_1.fastq SRR2584866_1.fastq
Output Summary (Cutadapt 4.9, Python 3.12.9):
Processing single-end reads on 1 core ...
Done 00:00:07 2,768,398 reads @ 2.8 µs/read; 21.79 M reads/minute
Finished in 7.626 s (2.755 µs/read; 21.78 M reads/minute).
=== Summary ===
Total reads processed: 2,768,398
Reads with adapters: 683,198 (24.7%)
Reads written (passing filters): 2,768,398 (100.0%)
Total basepairs processed: 415,259,700 bp
Total written (filtered): 377,219,215 bp (90.8%)
Impact
After adapter trimming, run FASTQC again to check the quality of the results.
$ module spider FASTQC
$ module load fastqc/0.12.1
$ module list # check what’s loaded (did you remember to `module purge` first?)
$ mkdir fastqc-out-trimmed
$ fastqc -t 4 -o fastqc-out-trimmed ecoli-fastq/SRR2584866-trimmed_1.fastq
Before trimming:

After trimming:

As you can see, adapter sequences have been successfully removed.