Assembly Construction
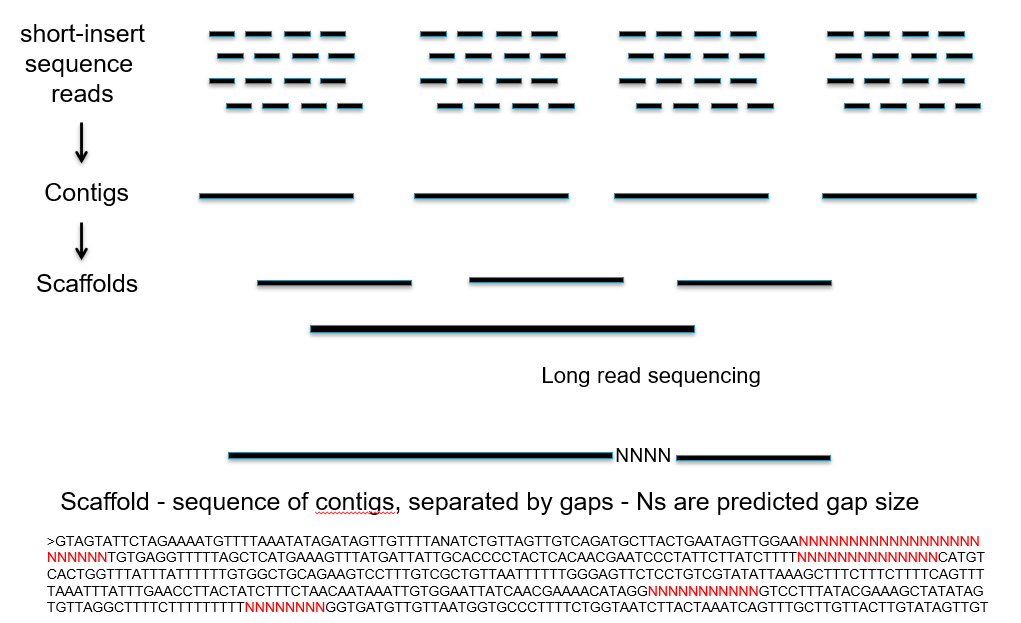
The construction of a sequence begins with short-insert sequence reads. These short reads are then combined to form longer, contiguous sequences called contigs. Contigs are then combined to form even larger scaffolds, though there often gaps between the contigs where the sequence is unknown. Long-read sequencing technologies can help order contigs in a sequence, as well as estimate the size of gaps (represented by the string of Ns).
Assembly Construction - Long Reads
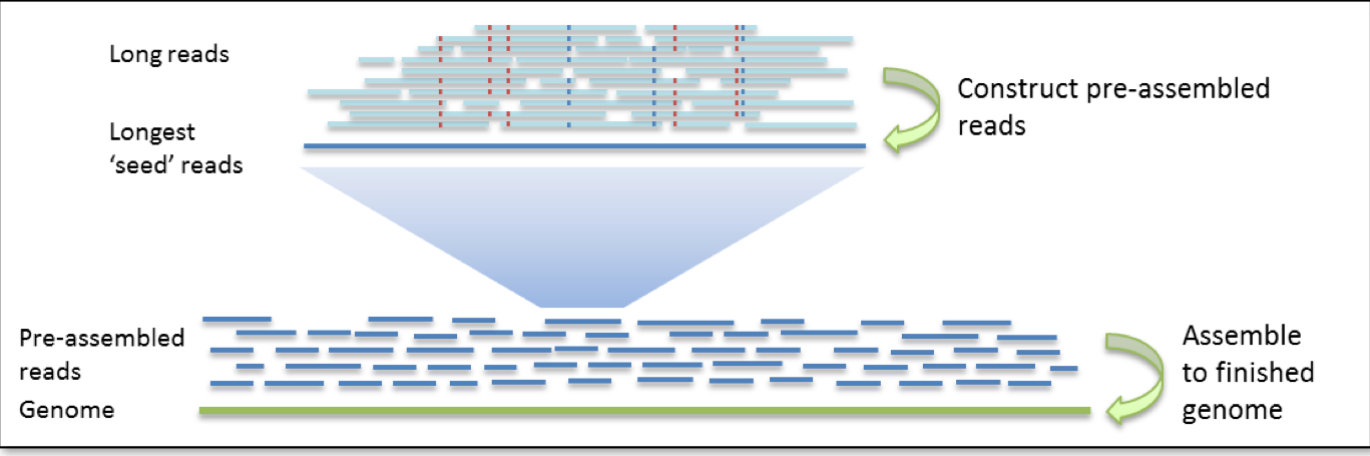
The assembly construction for long reads is still hierarchical, but proceeds in two rounds. The first round of assembly involves the selection of seed reads, or the longest reads in the dataset (with a user-defined length cutoff). In the second round, all shorter reads are aligned to the seed reads, in order to generate consensus sequences with high accuracy. We refer to these as pre-assembled reads, but they can also be thought of as “error corrected” reads. During the pre-assembly process, seed reads may be split or trimmed at regions of low read coverage (with a user-defined min cov for FALCON sense option). The performance of the pre-assembly process is captured in the pre-assembly stats file.
Repetitive Regions
Repetitive regions of DNA are often challenging during genome sequencing and assembly, as it is difficult to figure out where a section belongs in the overall sequence. Over 50% of mammalian genomes are repetitive, and large plant genomes tend to be even worse. For example, the Arabidopsis thaliana plant has a relatively small genome, yet 10% of its genome is made up of repetitive regions.
There are different categories of repetitive regions:
- SINEs - Short Interspersed Nuclear Elements
- LINES - Long Interspersed Nuclear Elements
- LTR - Long Terminal Repeats, retrotransposons
- Segmental duplications
- Low-complexity - microsatellites or homopolymers