Sequence File Formats

There are many different file formats that can be used for storing info about DNA sequences. With file formats, the format name usually denotes the suffix as well.
| Format Name | Suffix | Info |
|---|---|---|
| FASTA | .fasta, .fna, .fa | A standard file format |
| FASTQ | .fastq | Like FASTA, but also stores quality scores |
| SAM/BAM | .sam/.bam | Stands for Sequence Alignment Map, developed for NGS data and stores alignment information |
| VCF | .vcf | Stands for Variant Call Format |
| GFF3 | .gff3 | Stands for Generic Feature Format ver. 3 |
| GTF | .gtf | Stands for Gene Transfer Format, similar to GFF3 but contains additional gene annotation info |
FASTA
Here is an example of what a FASTA file looks like after the first time an assembly is opened.

FASTQ
Most modern sequencing platforms perform base calling and then return that data in FASTQ format, with quality scores included. (These quality scores are encoded using ASCII characters.) The FASTQ data can then be used for quality control and trimming.


SAM/BAM
Both SAM (Sequence Alignment Map) and BAM are file formats for sequence alignment files. These sequence alignment files provide context for raw data. Each file has eleven columns (tab-delimited), and one alignment is recorded for each line. SAM is a plain-text format (human readable), while BAM is a binary format. SAM/BAM files are often used with SAMTools (a suite of utilities for SAM/BAM files) and Picard (a collection of tools for sequencing data).
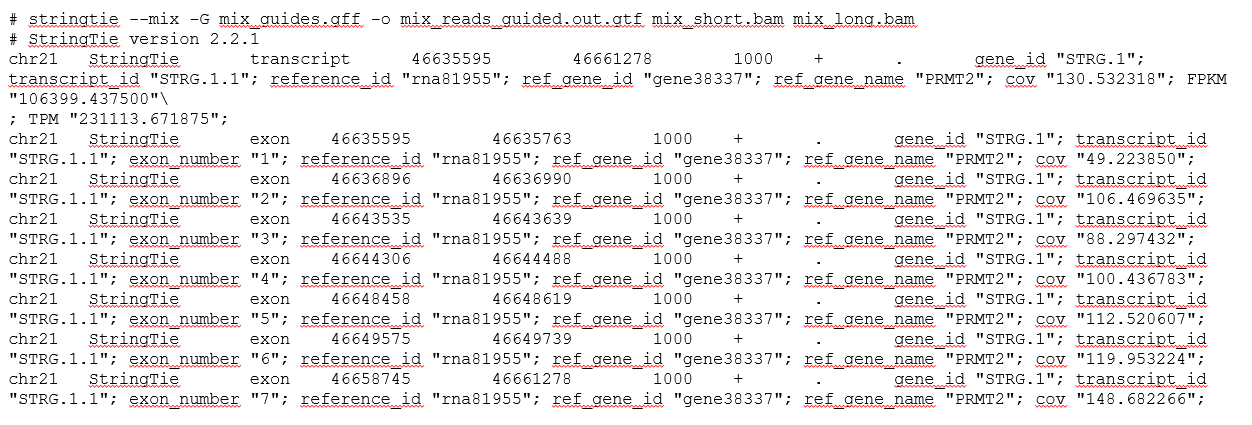
GTF
GTF (Gene Transfer Format) is a common format for annotating gene info on a genome.
References
- https://samtools.github.io/hts-specs/VCFv4.2.pdf
- https://github.com/The-Sequence-Ontology/Specifications/blob/master/gff3.md
- http://mblab.wustl.edu/GTF22.html
- https://help.basespace.illumina.com/files-used-by-basespace/quality-scores
- http://samtools.sourceforge.net
- https://broadinstitute.github.io/picard/