BUSCO
BUSCO (Benchmarking Universal Single-Copy Orthologs) is a tool used to assess the completeness of genome assemblies and annotations. It does this by looking for single-copy orthologs in the DNA sequence.
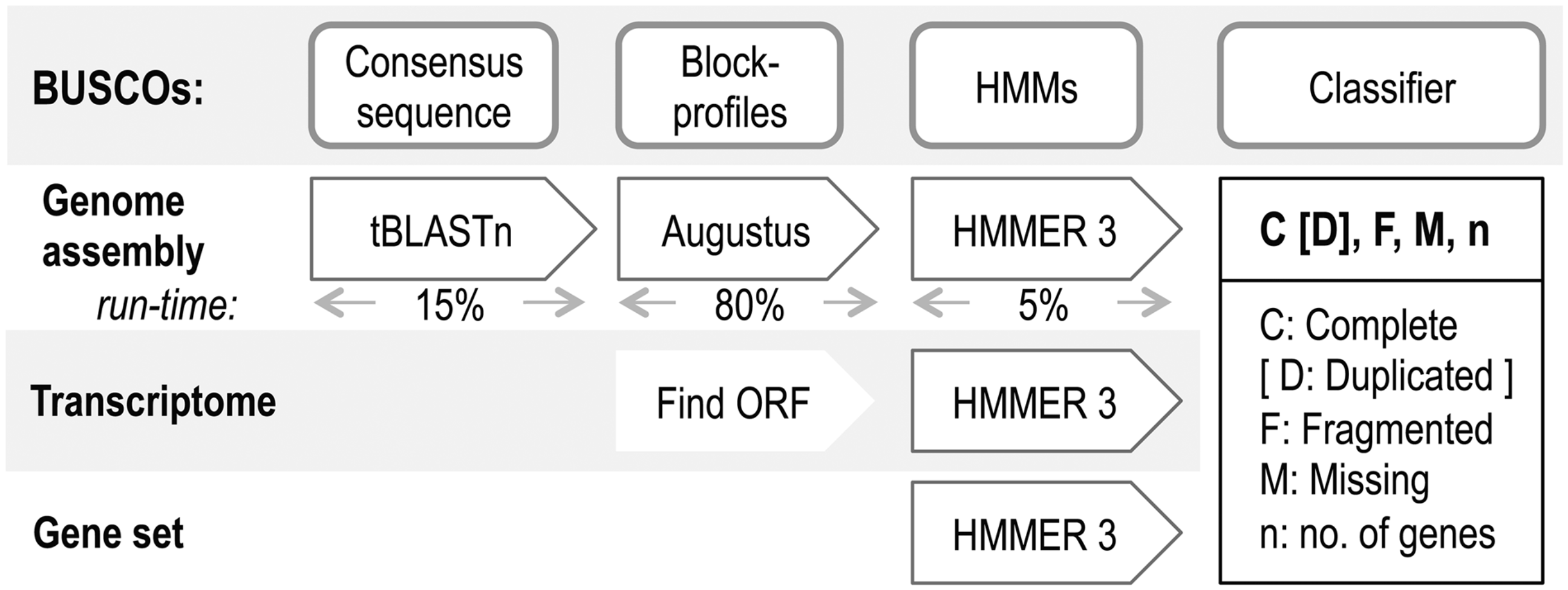
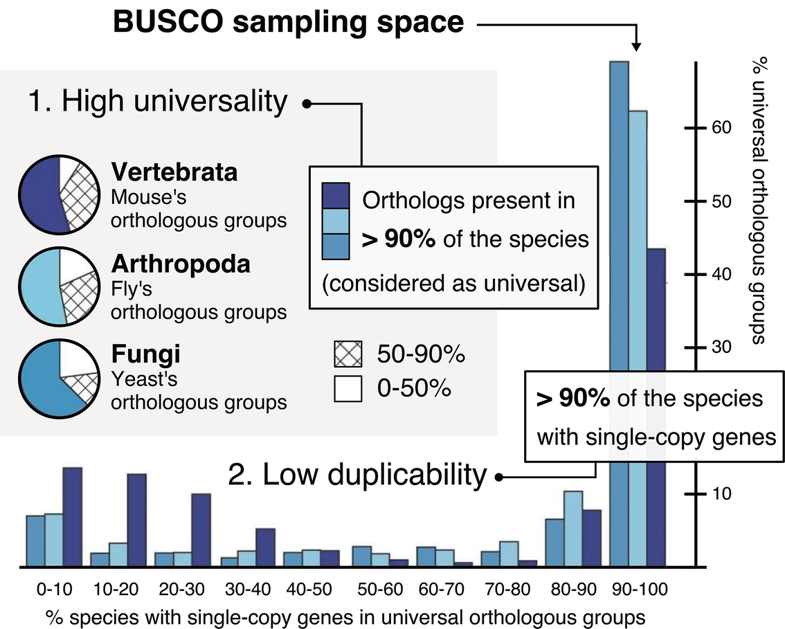
Running Busco Interactively
Running BUSCO interactively is good for testing.
To start an interactive session, use the ijob command:
ijob -c 1 -A hpc_training -p standard -v
To prepare to copy the test files, first move into your desired directory with the cd command:
cd /yourdirectory
Use the pwd command to check that you are in your desired directory:
pwd
Then copy the test files using the cp command (the dot at the end of the command means that the destination of the copied files will be the directory you are currently in):
cp /project/rivanna-training/genomics-hpc .
To check that all the files transferred correctly:
ls -lh
Now we can begin to load BUSCO. To check what versions of BUSCO are available on our system, run:
module spider busco
For this example, we will load version 5.8.2:
module load busco/5.8.2
To verify that the module was loaded, run:
module list
To actually run BUSCO on our Cyglarus-subset.fasta file, enter the command:
busco -i Cyglarus-subset.fasta -o Lep-Blue_out -m genome -l lepidoptera_odb10
The -l option indicates the lineage dataset used. This dataset will be downloaded automatically the first time you use it. The -o option will make a new output directory, or it will throw an error if the directory name already exists.
Running Busco with a Slurm Script
Slurm is a resource manager that can be used to run your code for you. Below is a Slurm script called busco_slurm_submit.sh.
#!/bin/bash
#SBATCH -A hpc_training # account name (--account)
#SBATCH -p standard # partition/queue (--partition)
#SBATCH --nodes=1 # number of nodes
#SBATCH --ntasks=1 # 1 task – how many copies of code to run
#SBATCH --cpus-per-task=4 # total cores per task – for multithreaded code
##SBATCH --mem=3200 # total memory (Mb)
#SBATCH -t 01:00:00 # time limit: 1-hour
#SBATCH -J busco-test # job name
#SBATCH -o busco-test-%A.out # output file
#SBATCH -e busco-test-%A.err # error file
#SBATCH --mail-user=yourid@virginia.edu # where to send email alerts
#SBATCH --mail-type=ALL # receive email when starts/stops/fails
module purge # good practice to purge all modules
module load busco/5.8.2
cd /project/rivanna-training/genomics-hpc/busco
# make new output directory or rename each time or throws an error if already exists!
busco -i Cyglarus-subset.fasta -o Lep-Blue_out -m genome -l lepidoptera_odb10
To submit the Slurm script and start a Slurm job, enter:
sbatch busco_slurm_submit.sh
When you run a Slurm script with the #SBATCH --mail-user=yourid@virginia.edu line and #SBATCH --mail-type=ALL line, Slurm will send you email notifications when your job starts, stops, or fails.
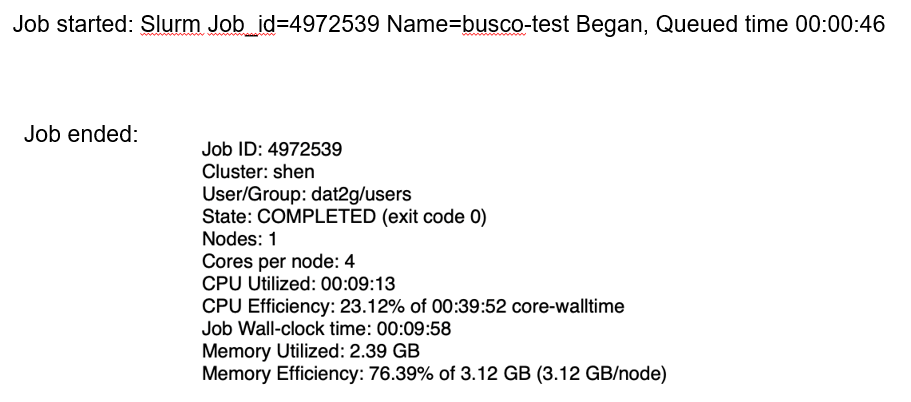
To immediately monitor the status of your Slurm job, enter:
squeue -u user_id
PD means pending, R means running, and CG means exiting.
Overall, the Slurm Script method should result in the same output files as when run interactively.
N50 Size
BUSCO is used to measure the completeness of a genome assembly. To measure the contiguity of a genome assembly (how continuous or unbroken the sequences are), you can use the N50 metric.
To calculate the N50 value for a genome, we place our contigs from largest to smallest on the genome. We then start with the longest contig and add the lengths of the next configs until the cumulative length reaches at least 50% of the total genome size. The length of the shortest contig added is the N50 value.
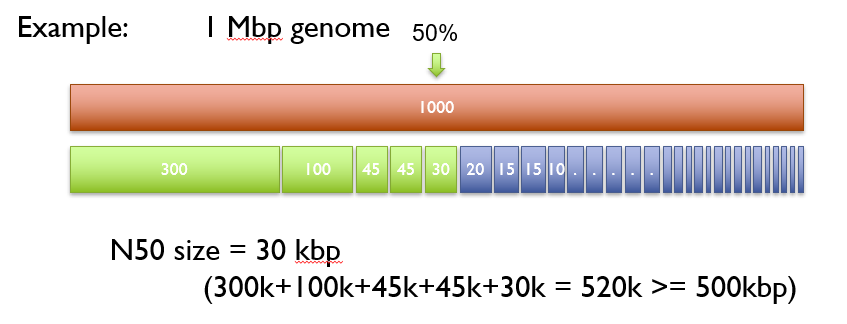
The N50 value is then compared with N50 values from genomes of similar size. A greater N50 is usually a sign of assembly improvement. Keep in mind that genome composition can bias comparisons. Another metric used alongside N50 is L50, which is the number of contigs that make up the N50. For example, a highly-contiguous assembly will have a high N50 value and a low L50 value.
Resources
- Bioinformatics. 2015;31(19):3210-3212. doi:10.1093/bioinformatics/btv351
- https://busco.ezlab.org