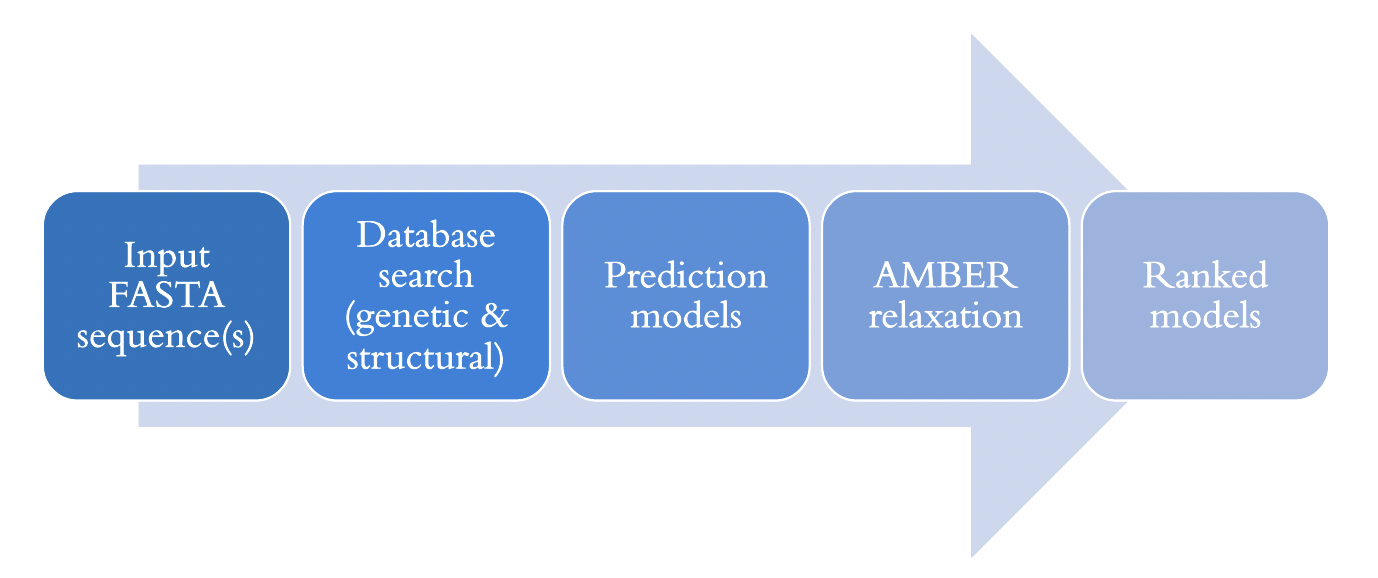
Pipeline and Architecture
-
User Input (FASTA)
The user provides a FASTA file: one sequence for monomer predictions or multiple sequences for multimer predictions. -
Database Search (Genetic & Structural)
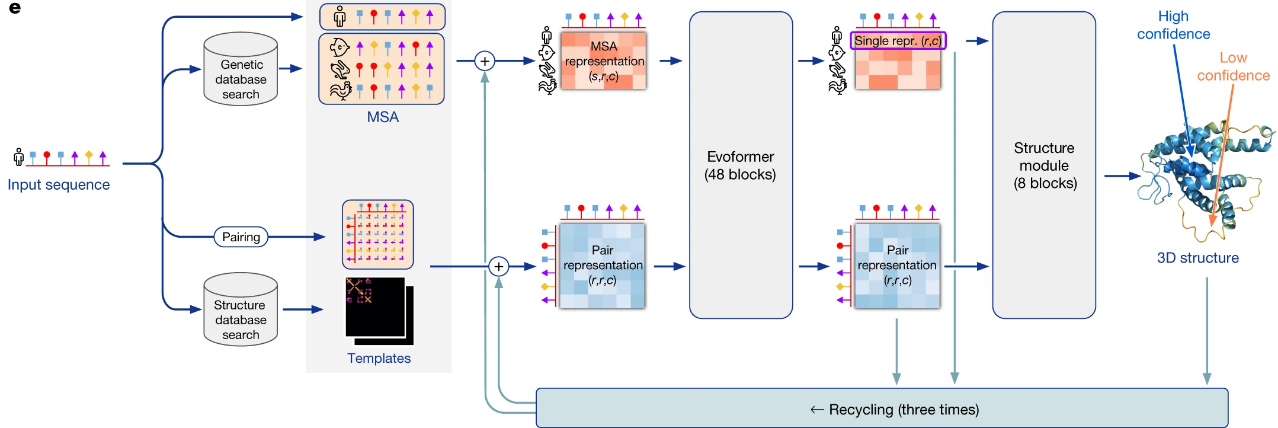
AlphaFold performs a multiple sequence alignment (MSA) using three tools:- JackHMMER on the MGnify database (top 5,000 matches retained),
- UniRef90 (top 10,000 matches retained),
- HHblits on UniClust30 and BFD (all matches retained).
A deep MSA is essential for accurate predictions; models show significant accuracy loss if fewer than 30 sequences are found.
In addition, AlphaFold performs a template search using HHSearch on the PDB70 database.
The top 4 templates are selected to initialize the prediction models. -
Prediction Models
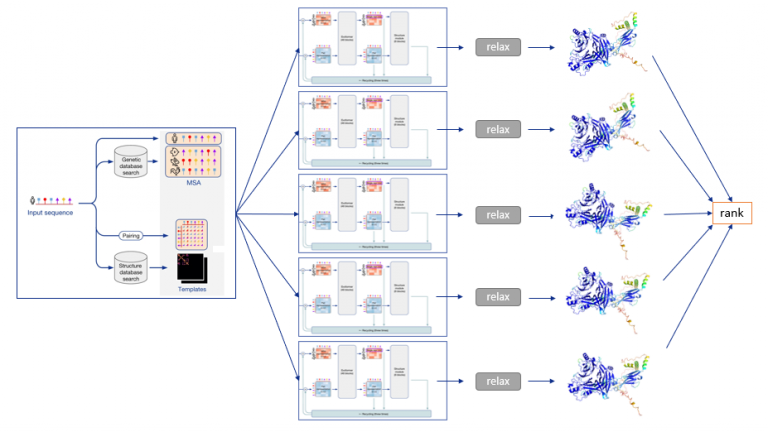
The MSA and structural templates are passed to five AlphaFold models with identical architecture but different parameter sets, each based on different randomization seeds. These five models share the same architecture but use different parameters, each initialized from a separate randomization seed.The model architecture includes:
- Evoformer blocks, which perform pairwise updates on the numerical MSA representation and a 2D amino acid pair representation, and
- Structure module blocks, which generate the folded 3D structure.
These modules are iteratively recycled three times, meaning the output structure from one iteration is reused as a template for the next.
The Evoformer and Structure modules are run in parallel across the five models, generating five distinct predictions. Each model output is then passed to relaxation and ranking steps independently.
-
AMBER Relaxation
The predicted structure is refined using AMBER relaxation, which eliminates side chain stereochemical violations (clashing side chains). The exact enforcement of peptide bond geometry is only achieved in the post-prediction relaxation of the structure by gradient descent in the Amber32 force field.- Empirically, this final relaxation does not improve the accuracy of the model as measured by the global distance test (GDT)33 or lDDT-Cα34, but does remove distracting stereochemical violations without the loss of accuracy.
-
Ranking
The five models are ranked based on a confidence score, and the highest-ranking structures are output as PDB files.